Startups and established pharma companies are keenly aware of the importance of patents and regulatory approvals in their drug development programs. Patents protect R&D investments, exclude competition and mitigate risk. Regulatory approval is necessary to market drug products. In the United States, both patents and FDA product approvals can provide exclusive rights for new pharmaceutical products.
The FDA’s product approvals ensure that marketed drugs are safe, efficacious and address unmet patient needs. The New Drug Applications (NDA) and Biologics License Application (BLA) are the two submission pathways available to sponsors for approving their new drugs. The two pathways differ from each other in terms of their allowed product categories, criteria for approval, and associated regulations. Marketing approval for small molecule drugs falls under NDA while all biological products must be submitted through the BLA application process.
The Center for Drug Evaluation and Research (CDER), FDA’s unit that is partly responsible for NDA and BLA applications, approves drug products such as small molecules and therapeutic antibodies after evaluating their safety and efficacy. Some of these products are classified as novel as they are new molecular entities (NMEs) and new therapeutic biological products. Novel products have active ingredients that have never been marketed in the United States. Other original approvals come in the form of new or expanded use, dosage forms, formulations and biosimilars.
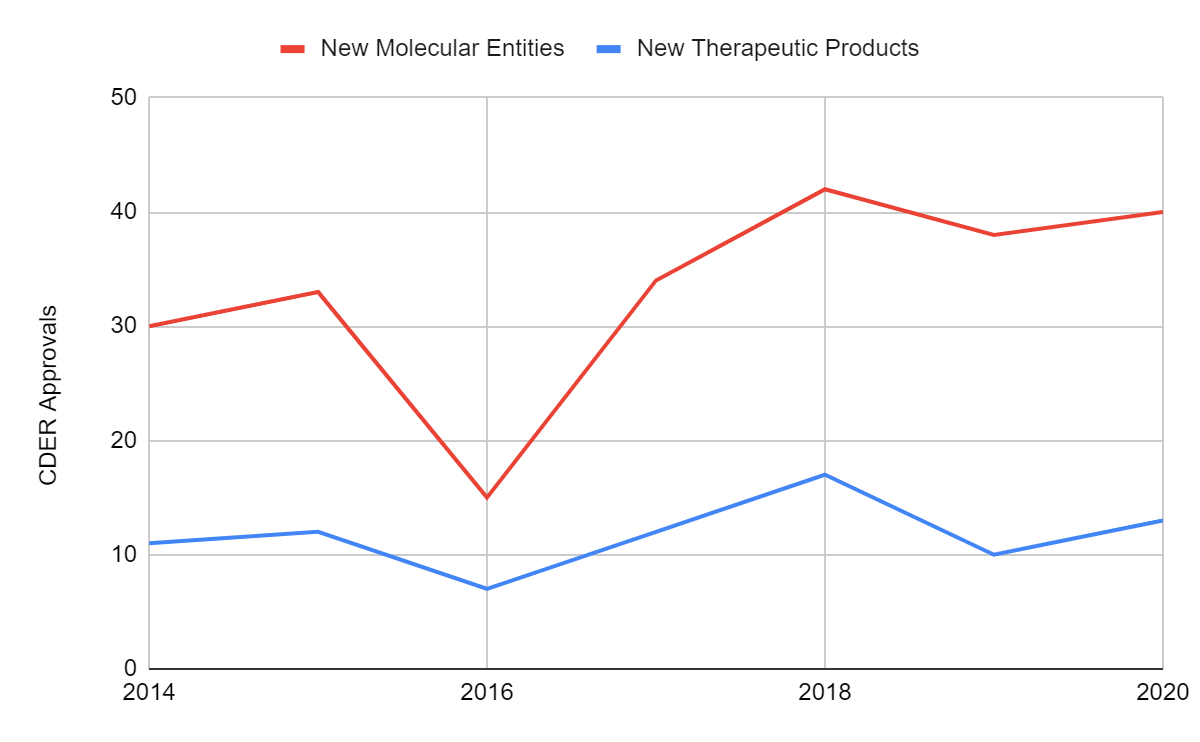
The above chart shows novel approvals for NMEs and new therapeutic biological products under the CDER for the 2014-2020 period. The CDER approved a record number of new drugs in 2020 in spite of COVID-19 challenges. Forty NMEs were approved under NDAs while the remaining 13 were new biological therapeutics approved under BLAs. The combined 53 approvals is only 6 short of the FDA’s all-time high number of 59 approvals in 2018. The upward trend in novel drug approvals is likely to continue into 2021. Thirty-three NMEs and four new biological therapeutics have been approved between January 01, 2021 – May 21, 2021.
Below, we shall discuss some of the recent BLA holders and their related patents and patent applications.
GlaxoSmithKline
GlaxoSmithKline’s (GSK) Jemperli is a programmed death receptor-1 (PD-1) blocking monoclonal antibody (dostarlimab-gxly). It was approved on April 22, 2021 under the accelerated pathway. The treatment is for mismatch repair-deficient (dMMR) recurrent or advanced endometrial cancer patients who have progressed on to or have received prior platinum-containing regimen treatment. The antibody was initially developed by AnaptysBio and later licensed to Tesaro, which was acquired by GSK in 2019 for $5.1 billion.
Dostarlimab will compete with the category leader Pembrolizumab (KEYTRUDA; Merck). Pemrbrolizumab is currently approved for use in endometrial cancers in combination with lenvatinib (LENVIMA; Eisai). GSK will also look to dostarlimab to compete in other indications, such as solid cancers.
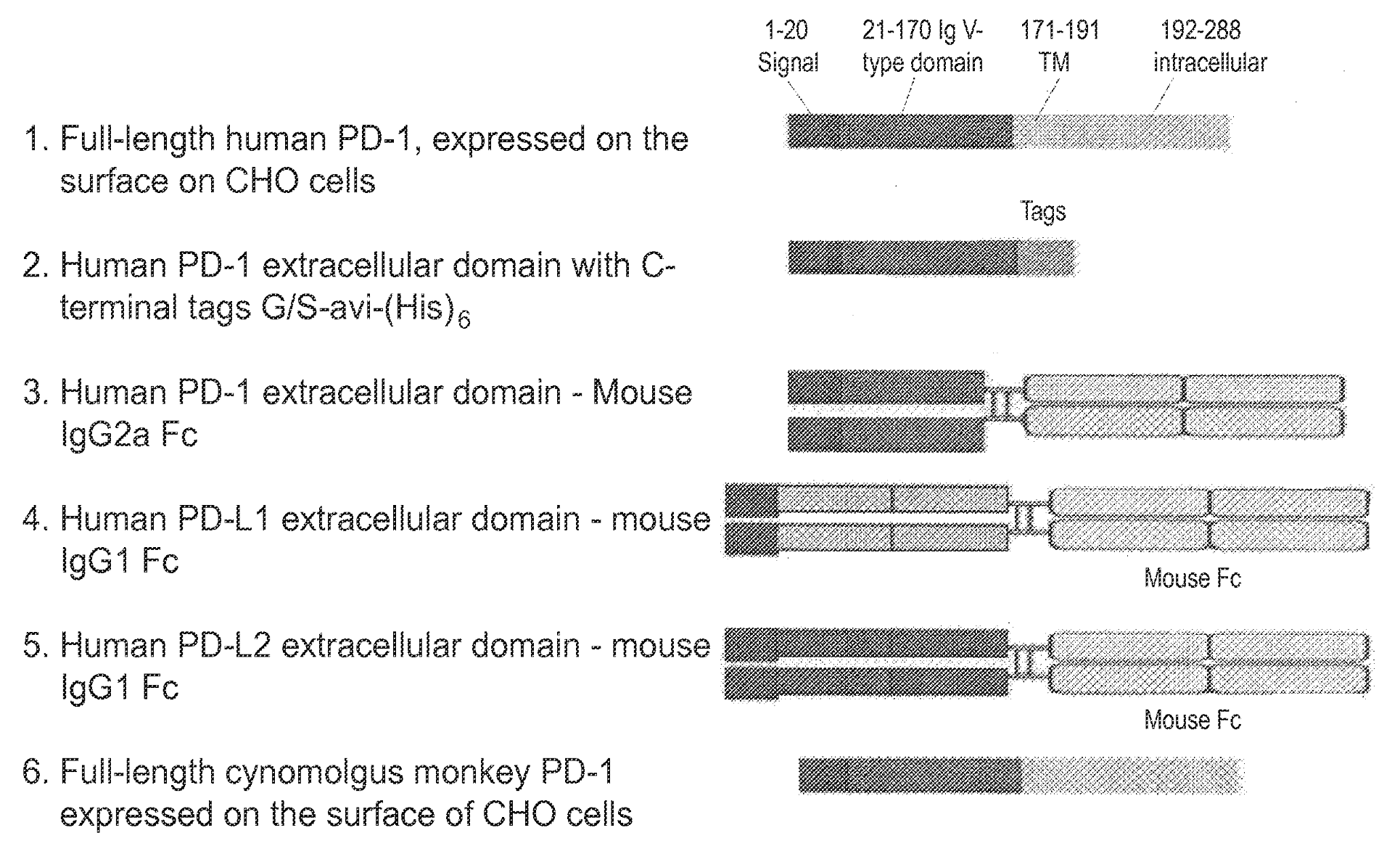
US9815897B2 titled “Antibodies Directed Against Programmed Death-1 (PD-1)” assigned to AnaptysBio describes a programmed death-1 (PD-1) binding antibody, related vectors, and compositions, and for treating cancer or an infectious disease.
ADC Therapeutics
ADC Therapeutics focuses on antibody-drug conjugates (ADC), treatments that combine the selectivity of antibodies with the efficacy of small molecule drugs. It received its first FDA approval on April 23, 2021. Zynlonta (loncastuximab tesirine) was approved under an accelerated pathway for the treatment of patients with relapsed or refractory (r/r) diffuse large B-cell lymphoma (DLBCL). The ADC is a humanized monoclonal antibody that binds to human CD19 conjugated to a pyrrolobenzodiazepine (PBD)-based cytotoxic warhead through a linker, a short peptide sequence used for the attachment of the anti-cancer PBD molecule with the antibody.
The single-agent treatment offers an alternative to existing CD-19 targeted CAR-T therapies in DLBCL. With this approval, the company joins a small group of companies such as Pfizer, Roche/Genentech, and AstraZeneca with approved ADC products.
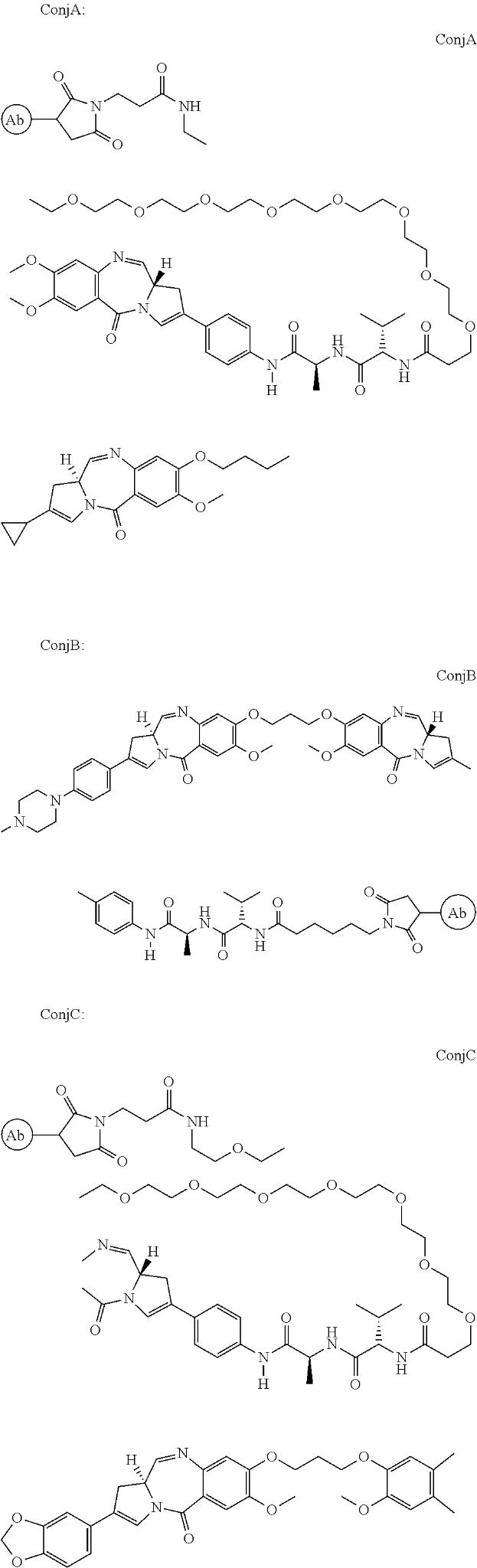
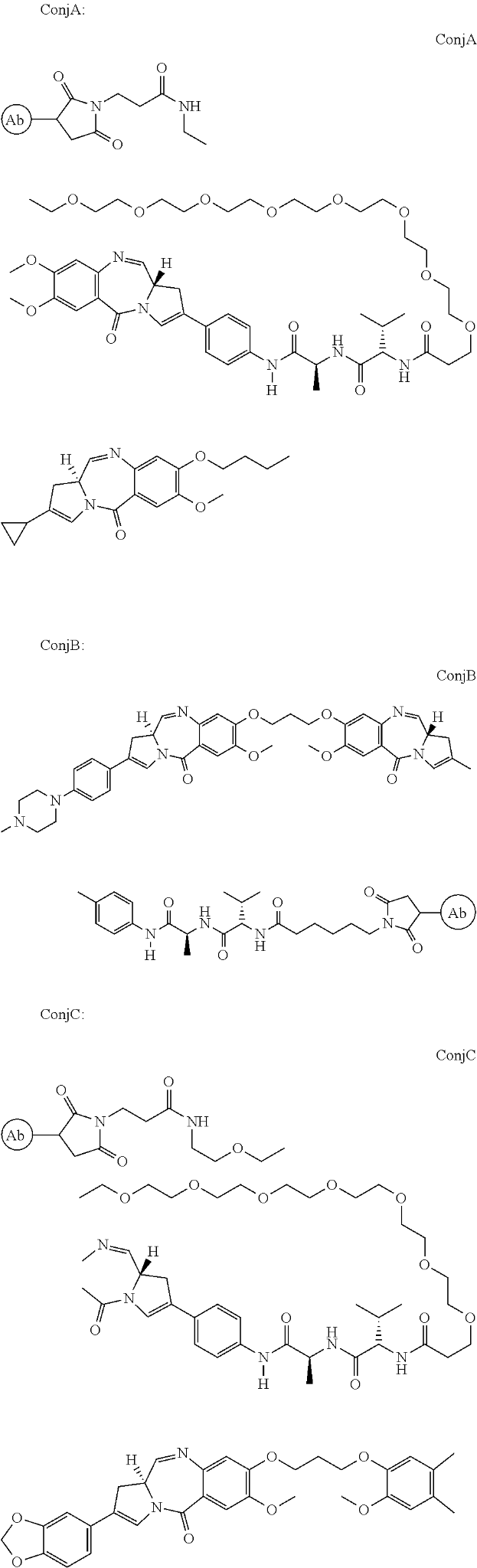
ADC Therapeutics’ patent, US9931414B2, titled “Pyrrolobenzodiazepine-antibody Conjugates” describes conjugates of PBD that binds to CD19 antibody. Pyrrolobenzodiazepines (PBDs) have the ability to recognise and bond to specific sequences of DNA. The anti-CD19 attachment is suitable for delivering the PBD to a preferred site such as a cancer that overexpressed the CD19 receptor. The conjugate allows the release of an active PBD compound that does not retain any part of the linker. The ADC may be administered to the patient as a combination with an existing therapy such as rituximab.
Regeneron Pharmaceuticals
Regeneron’s EVKEEZA (evinacumab-dgnb) is an injectable prescription medicine used as an adjunct with other low-density lipoprotein (LDL) lowering medicines in people older than 12 years of age with a rare type of high cholesterol called homozygous familial hypercholesterolemia (HoFH). Familial hypercholestrolaemia (FH) is an inherited genetic condition where the patient’s cholesterol levels are higher than normal from birth as their liver is unable to break down or remove excess cholesterol, specifically LDL-C. As a result, the FH population is susceptible to heart disease at a relatively young age.
The drug was approved by the FDA on February 11, 2021 under breakthrough designation. The drug is also classified under Orphan status for treating a rare disease or condition. It is a fully human monoclonal antibody that binds to and blocks the function of angioprotein-like protein 3 (ANGPTL3) thereby reducing LDL-C levels. The new class of biologic will compete with other lipid-lowering therapies, such as the PCSK9 inhibitor candidates: Regeneron/Sanofi’s Praulent (alirocumab), Amgen’s Repatha (evolocumab) and Novartis’s approval-pending Leqvio (inclisiran). Regeneron is also studying the drug in refractory hypercholesterolemia and severe hypertriglyceridemia.
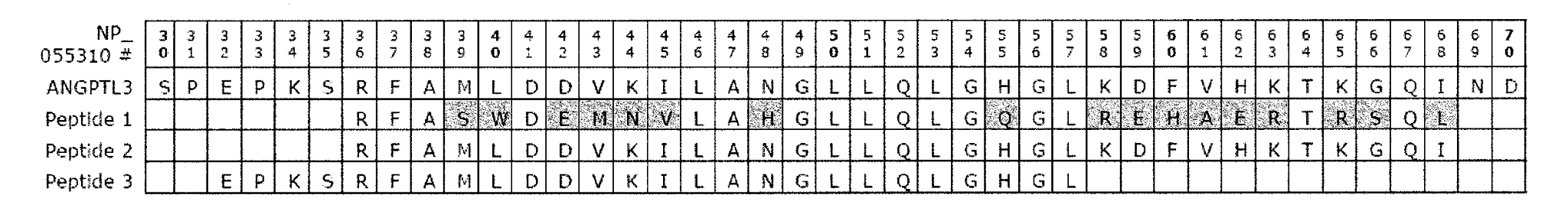
US9018356B2 titled “Anti-ANGPTL3 antibodies and uses thereof”, and US20200061189A1 titled “Methods for Treating Patients with Familial Hypercholesterolemia” each describe a fully human antibody that acts against human angiopoietin-like protein 3 (hANGPTL3) for treating diseases or disorders, such as hyperlipidemia, hyperlipoproteinemia and dyslipidemia, hypertriglyceridemia, hypercholesterolemia, chylomicronemia, atherosclerosis, etc.
Johnson & Johnson
Janssen (a subsidiary of J&J) received a nod for its Rybrevant (amivantamab-vmjw) on May 21, 2021. It is a fully human bispecific antibody indicated for treating adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations following platinum chemotherapy.
It focuses on a hard-to-treat subset of NSCLC patients as determined by the accompanying FDA-approved companion diagnostic test from Guardant Health’s Guardant360 CDx liquid biopsy blood test. The antibody is designed to target a combination of EGFR and hepatocyte growth factor receptors (MET) while existing tyrosine kinase inhibitors target aberrant EGFR signaling from the inside. The breakthrough targeted therapy gives J&J access to a niche market of 2-3 % of NSCLC patients. J&J will also look to expand its use of other indications, such as other EFGR-mutated cancers.

US9580508B2 titled “Bispecific EGFR/c-Met Antibodies” and US9695228B2 titled “EGFR and c-Met Fibronectin Type III Domain Binding Molecules” describe an isolated bispecific epidermal growth factor receptor (EGFR)/hepatocyte growth factor receptor (c-Met) antibody, such as bispecific fibronectin type III (FN3) domain that specifically binds epidermal growth factor receptor (EGFR) and hepatocyte growth factor receptor (c-Met), and blocks binding of ligands to the receptors. The antibodies are used for treating cancers such as non-small cell lung cancer (NSCLC). Mutations can be located in any portion of an EGFR gene or regulatory region associated with an EGFR gene and include mutations in exons 18, 19, 20 or 21, or mutations in the kinase domain.
Although patent information is included by the NDA holder as a part of their FDA submission, such a requirement is not currently in place for BLA holders. Even if such information is available, it does not inform of the NDA/BLA holder’s unlisted patents/pending applications and the therapeutic landscape. A comprehensive patent search can reveal the full extent of infringement risk and competitor activity.
Know more about our Search Services.