The U.S. Food and Drug Administration (FDA) has been at the forefront in our fight against COVID-19. Immediately after the outbreak, the FDA initiated several measures for tackling the pandemic, such as granting expanded access and new approvals. Understandably, most of the COVID-19 related approvals have come under the Emergency Use Authorisation (EUA) pathway rather than the usual regulatory approval process. Such approvals have helped healthcare workers get access to critical COVID-19 medical device, such as in-vitro diagnostic kits, point of care devices, etc.
The filing of intellectual property (e.g. patents) and regulatory submissions are closely interlinked. Patent filings typically precede regulatory filings and aim to cover broad aspects of the technology. Regulatory submissions that do not fully consider intellectual property filings can have potential consequences regarding the patentability of a filing, and its validity/enforceability that could result in infringement proceedings1. We examine recent regulatory approvals at the FDA’s Center for Devices and Radiological Health (CDRH) and selected patent publications.
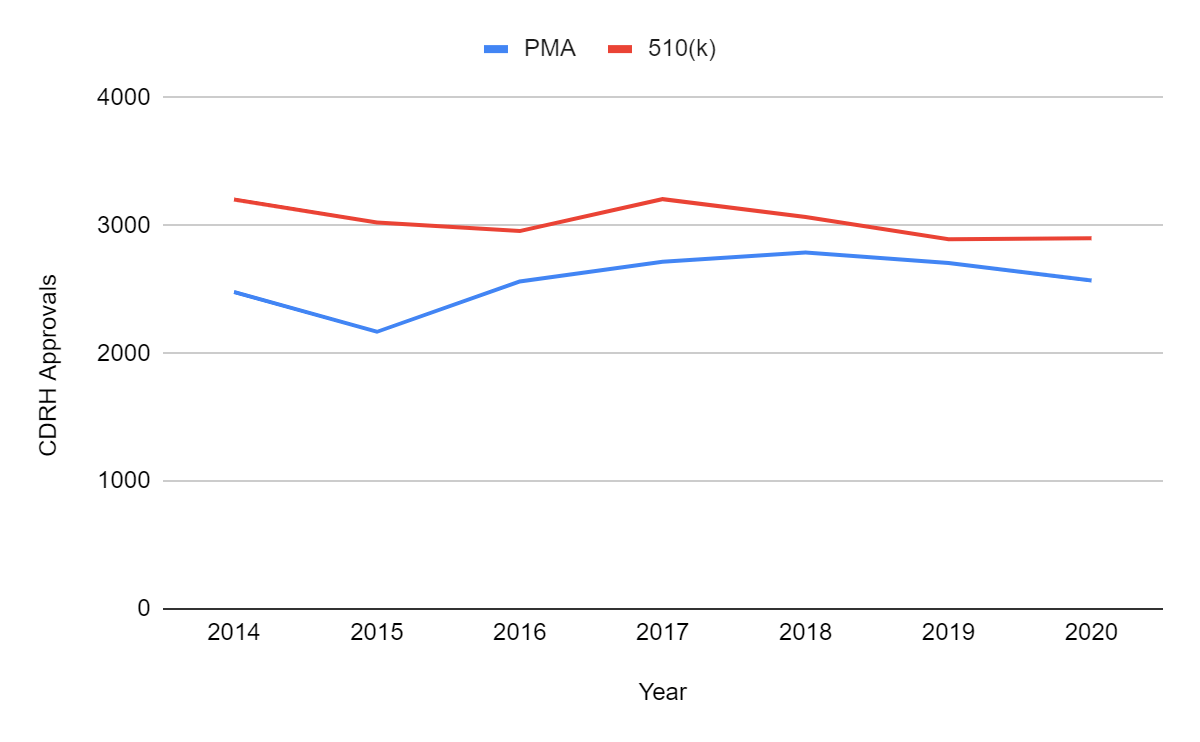
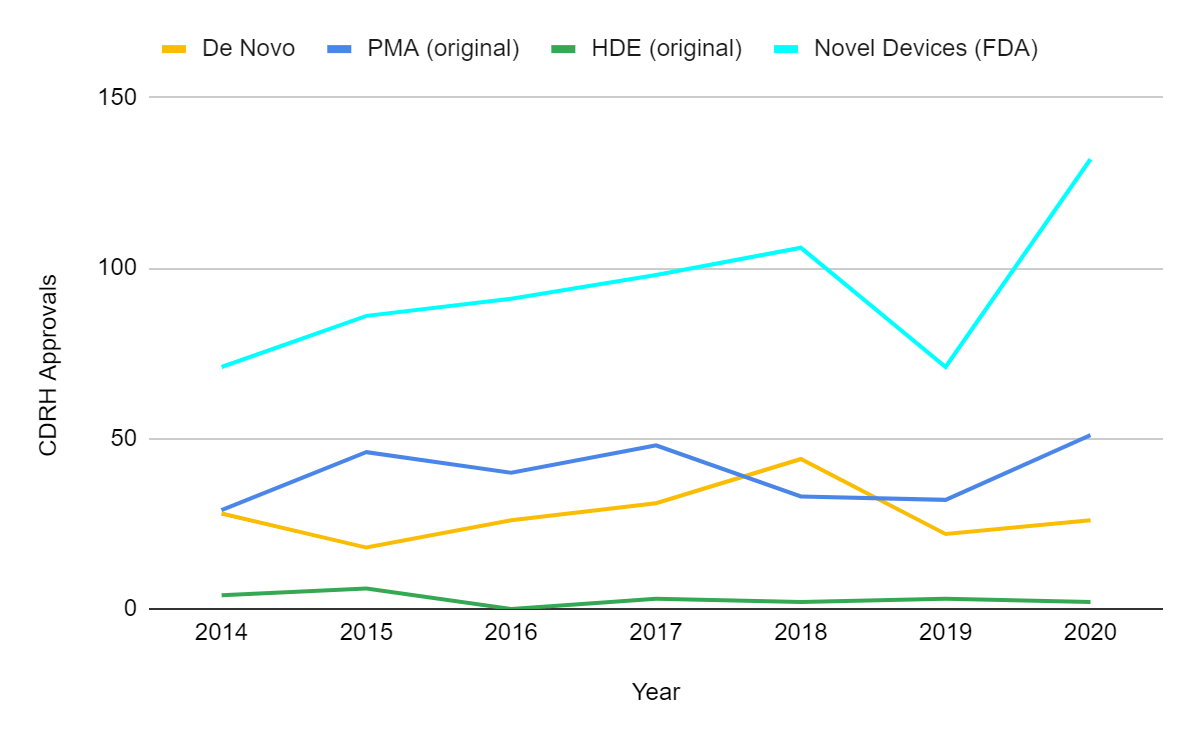
From the above, it is clear that FDA approvals did not decrease in 2020 in spite of COVID-19. There has been no major impact in the two major pathways administered by the Center for Devices and Radiological Health (CDRH): pre-market approvals (PMA) and premarket notification 510(k) clearances. There were 26 de novo and 2 humanitarian device exemption (HDE) authorized devices in 2020, the other pathways overseen by CDRH. As of May 1, 2021, there were 742 PMA approvals, 1039 clearances through 510(k), 12 de novo classifications and 1 HDE authorization suggesting that the stable trend will continue into 2021. It is an impressive feat by the FDA considering all the recent approvals related to COVID-19 have come through emergency use authorizations (EUAs).
Notably, the FDA authorized a record high 132 novel medical devices in 2020, surpassing the 40-year high mark set in 2018. The FDA considers applications in PMA, HDE, De Novo pathway, and a subset of 510(k) clearances and EUA approvals as “novel” devices2. This is because the FDA’s definition of “novel” or innovative does not simply mean “new.” The device has to address an unmet need or be safer or more effective than currently available alternatives.
Emergency Use Authorization (EUA)
The FDA issued 625 EUAs for medical devices in 2020 in response to the pandemic. These devices ranged from personal protective equipment, decontamination equipment, ventilators, in-vitro diagnostics, patient monitoring devices, respiratory assist devices, wearable or remote monitoring devices, infusion pumps, hemodialysis devices and other medical devices3.

Image Source: Cepheid
For example, Cepheid’s point-of-care (POC) COVID-19 diagnostic test – Cepheid Xpert Xpress SARS-CoV-2 test – was the first POC diagnostic device to be EUA approved in March 2020. The test is run on their rapid molecular diagnostic platform – GeneXpert System.
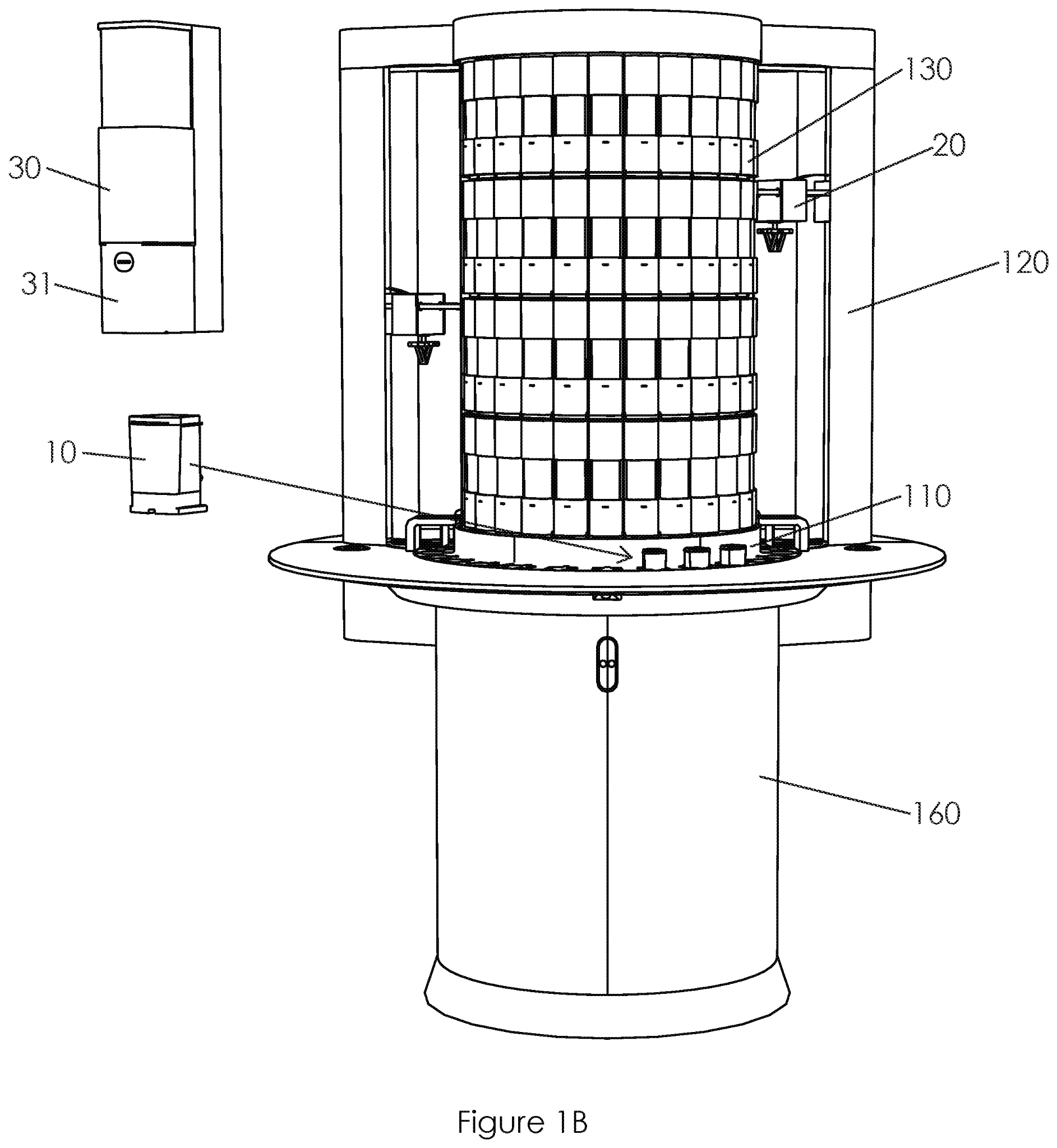
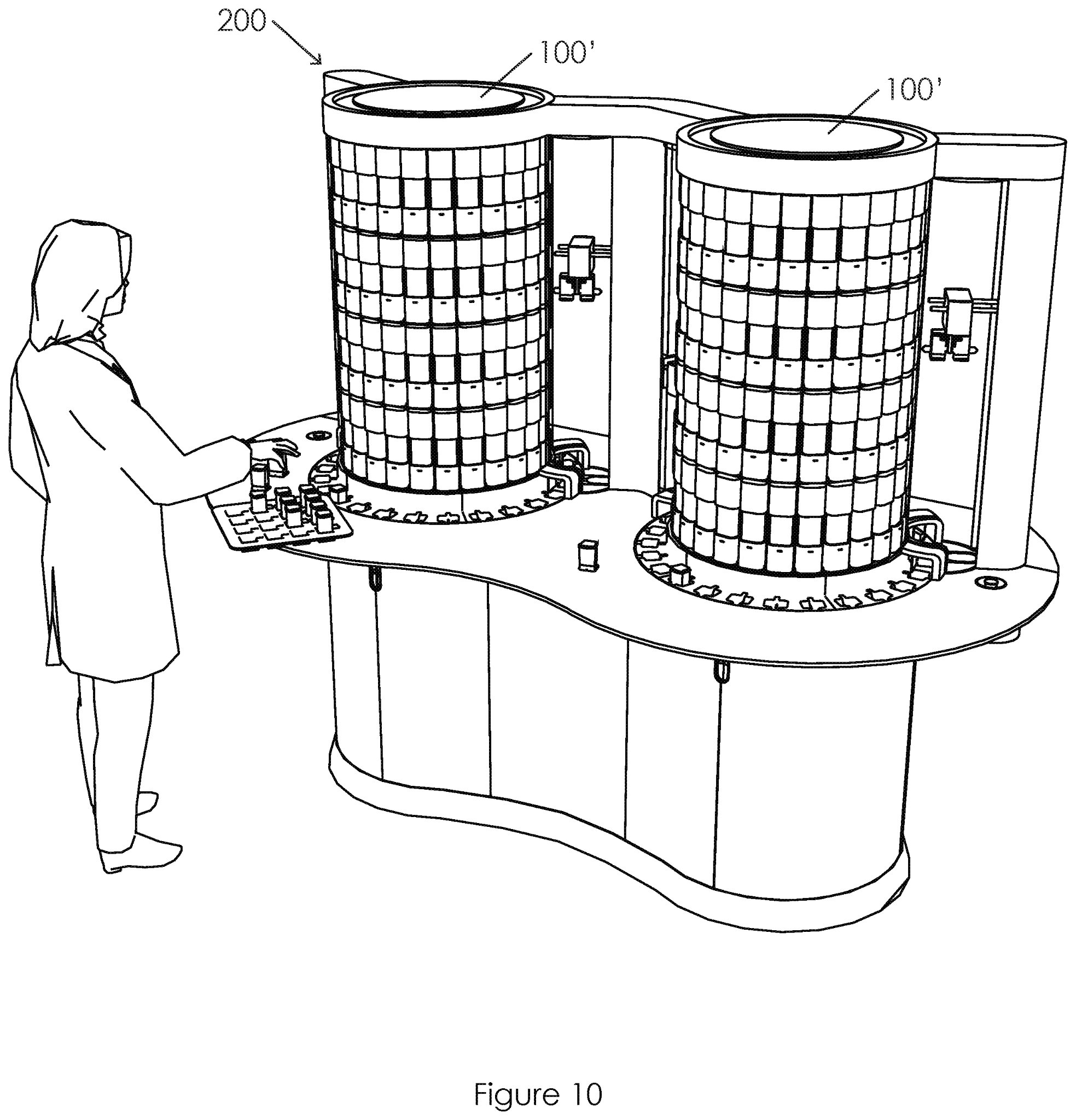
US20210072269A1 titled “Sample Processing Module Array Handling System and Methods” assigned to Cepheid claiming priority to Nov. 18, 2016 claims a method of handling a high throughput system processing large volumes of biological samples. The samples in a diagnostic assay cartridge (10) are placed using movement between the loader (20) and array support assembly for efficient processing. Specifically, the disclosure appears to be solving the problem of the need for a high throughput system that can be installed in existing facilities. The system is a cartridge-based system for nucleic acid amplification for running rapid diagnostic tests. Existing systems are large and expensive while smaller systems are not high-throughput systems resulting in unacceptable delays during a critical situation such as the pandemic.
Pre-Market Notification – 510(k)
Most Class I and some Class II devices are exempt from a 510(k) submission if they present a low to moderate risk to a patient. Premarket notification 510(k) for low to moderate risk devices is the most widely used pathway contributing to over 50% of all approvals before the CDRH. In order to get clearance through the 510(k) pathway, the applicant must demonstrate that the new device is substantially equivalent to a predicate device. A predicate device is an existing medical device that may be legally marketed in the U.S and used as a point of comparison for establishing equivalence. The predicate device can be from their existing product portfolio or from a third party’s portfolio.

Image Source: Roche
Roche Molecular Systems received a 510(k) clearance for a BK virus detection test that is run on their cobas 6800/8800 platform on January 29, 2021 (K203220). The clearance is for the quantitation of BK virus DNA from human plasma/urine and is similar in characteristics to the predicate (K202215; September 2, 2020), which itself traces its clearance to its predicate (DEN200015; July 30, 2020) for the quantitation of the Epstein-Barr Virus (Ebv) DNA. Roche also received other clearances and authorizations for its Cobas platform.
Interestingly, a recent PCT application WO2021013972A1 titled “Compositions and Methods for Detection of Epstein Barr Virus (EBV)” filed by Roche with priority to July 25, 2019, claims a method for rapidly detecting Epstein Barr virus in a sample by a PCR-based method involving an amplifying step, a hybridizing step, and a detecting step. Kits based on primers and probes for targeting the virus and kits are provided therein.
Pre-Market Approvals – PMA
PMA is the most stringent pathway for medical device marketing approval. Devices that require a PMA are Class III devices that pose a significant risk of illness or injury, or devices that are not eligible for the 510(k) process. Such class III devices are often novel and cannot become predicate devices by the 510(k) pathway. Pre-market approvals are based on a determination by the FDA that the application contains sufficient valid scientific evidence, such as clinical trial data, to assure that the device is safe and effective. As a result of its stringent requirements, PMA may take longer than 6 months for approval and is costly.
The original PMA approvals in 2020 were mostly in the area of diagnostics – a trend that has extended into 2021. For example, the top applicant DiaSorin received PMAs for diagnostic tests on its Liaison platform for detecting hepatitis B, hepatitis C, and HIV antibodies in a patient’s blood. The other major therapeutic areas were cardiology, ophthalmics, orthopedics, neurology and urology. Roche, Philips, Ventana Medical Systems, Medtronic, Abbott, Boston Scientific and Foundation Medicine were other notable applicants.
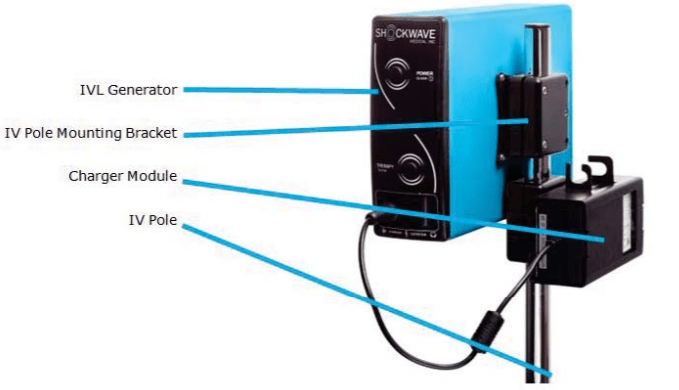


Image Source: FDA
Shockwave Medical received PMA approval through the Breakthrough Device program on February 12, 2021, for its Shockwave Intravascular Lithotripsy (IVL) System with Shockwave C2 Coronary Intravascular Lithotripsy (IVL) Catheter. The Coronary IVL System is designed to enhance stent outcomes by enabling delivery of the calcium disrupting capability of lithotripsy prior to balloon dilatation at low pressures. It consists of an IVL balloon catheter with two integrated pairs of lithotripsy emitters, a lithotripsy generator, and a connector cable. NCT03595176 evaluates the safety and effectiveness of the system in de novo, calcified, stenotic coronary arteries prior to stenting.
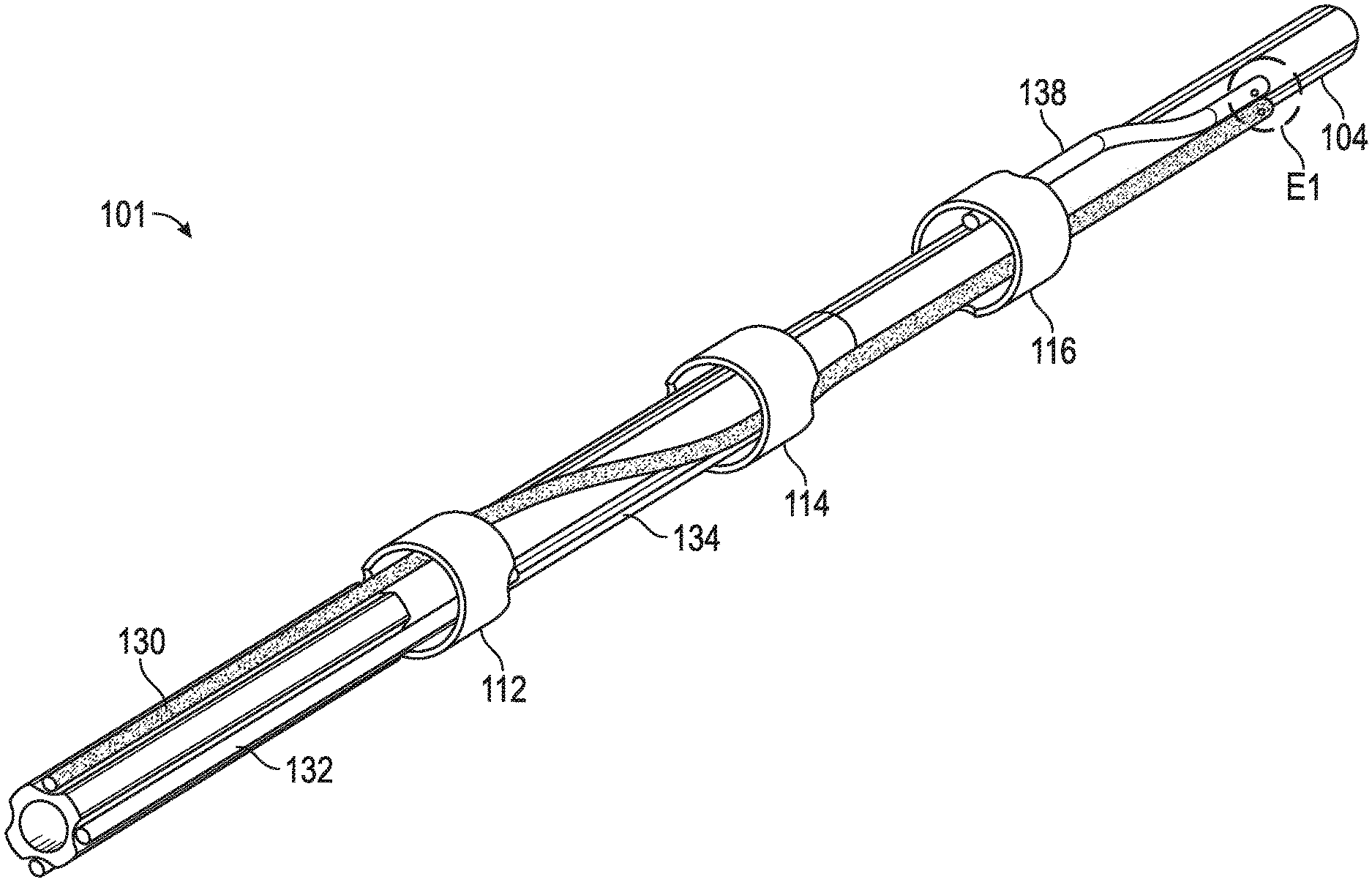
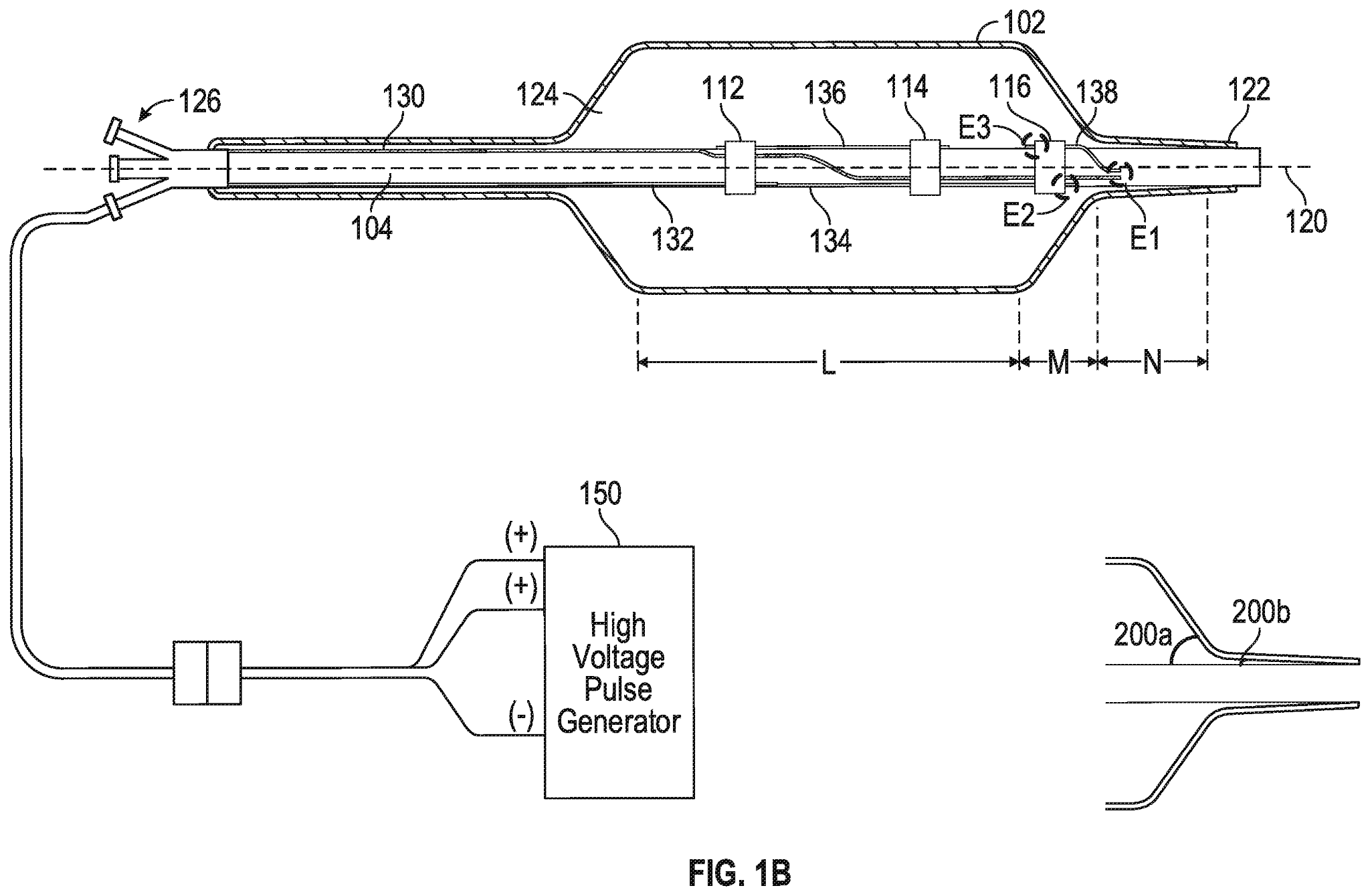
A recent patent application from Shockwave Medical – US20210085383A1 – titled “Low Profile Electrodes For a Shock Wave Catheter” claiming priority to September 24, 2019, describes a system for treating tight, hard-to-cross calcified lesions in which an angioplasty balloon is used to dilate the lesions and provide shock waves to restore normal blood flow in a patient’s artery. Such a device includes an elongated tube (104) and a balloon (102) wrapped circumferentially around the tube and sealed to a distal end of the tube. During treatment, the device is advanced into a patient’s vasculature and the balloon is inflated with a conductive fluid such that the balloon is fixed to walls of the vasculature proximal to the calcified lesion. The balloon includes at least one low-profile emitter (E1-E3) positioned near the distal end of the balloon, which may be activated to generate shock waves to break loose calcifications in the lesion. After calcium in the tight lesion has been modified, the balloon can be deflated and advanced further into the lesion to continue treatment.
De Novo Classification
The de novo process provides an alternative pathway for approval of novel medical devices for which there is no legally marketed predicate device. Such a request can be made after these devices are automatically placed in Class III and after receiving a “not substantially equivalent” (NSE) determination in response to a premarket notification [510(k)] submission. Alternatively, such a request can be made without first submitting a 510(k) and receiving an NSE determination. A De Novo classification request may result in these devices being classified into Class I or Class II for marketing purposes and allows them to become predicates for future premarket notification [510(k)] submissions.
There were twenty-six De Novo approvals in 2020. The approvals were related to diagnostics (8), gastroenterology/urology (6), cardiology (3) and orthopaedics (3). At the time of the writing of this article, there were already 12 De Novo approvals in 2021.

Image Source: FDA
The neurological device “Q-collar” was approved on February 26, 2021, under the De Novo classification. The device is an external compression device for internal jugular vein compression. It is in the form of a C-shaped collar that applies compressive force to the neck and increases blood volume to help reduce movement of the brain within the cranial space which may occur during head impacts. When worn around the neck during sports activities, the Q-Collar provides compressive force to the internal jugular veins, which in turn increases the blood volume in the skull’s blood vessels. Typically, when people experience blunt trauma accidents, the brain moves unrestrained in the skull, which is known as a “slosh.” The Q-Collar’s increase in blood volume in those blood vessels creates a tighter fit of the brain inside the skull and reduces the “slosh” movement. By reducing the movement of the brain within the cranial space, the Q-Collar may aid in the protection of the brain from the effects of head impacts.
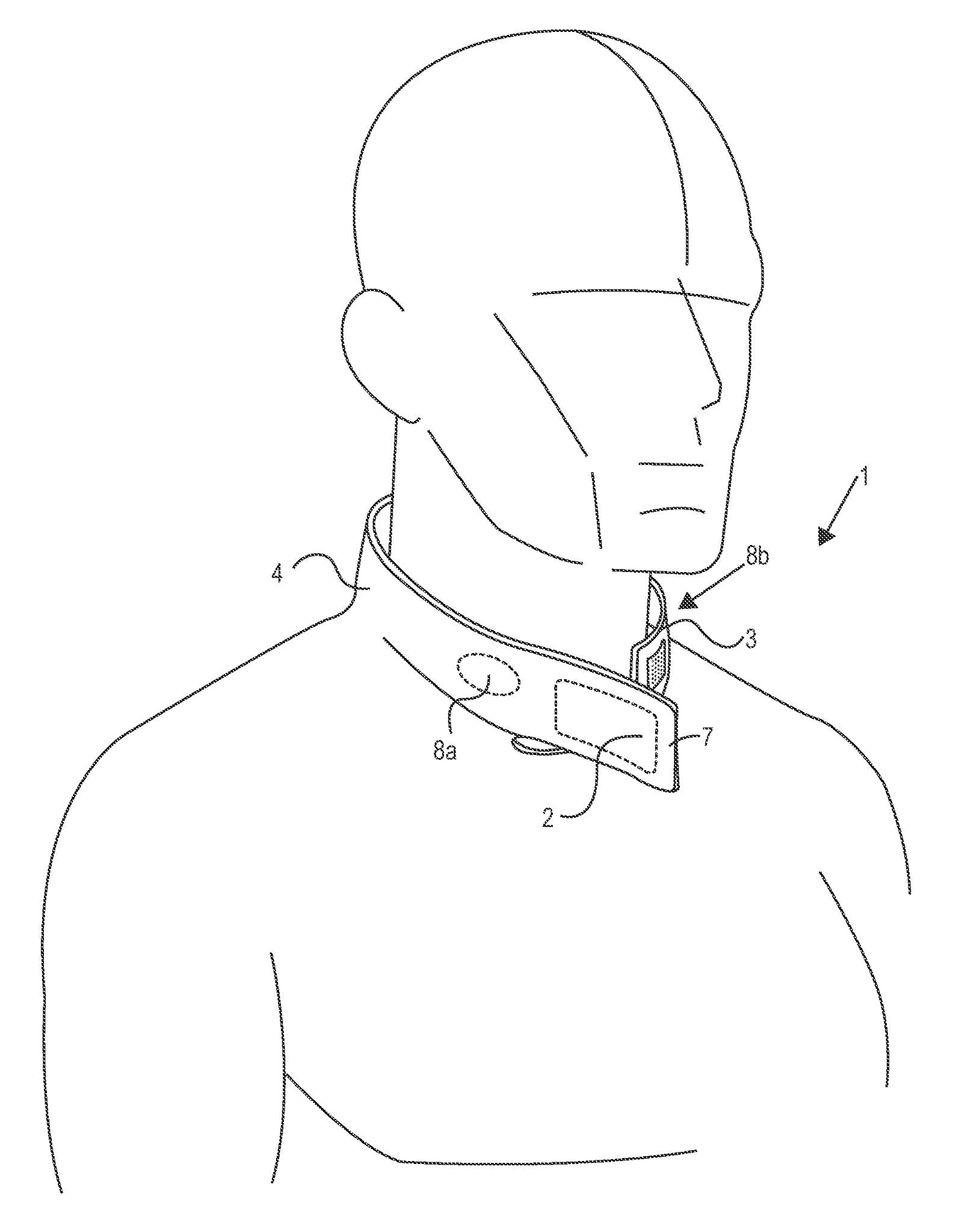
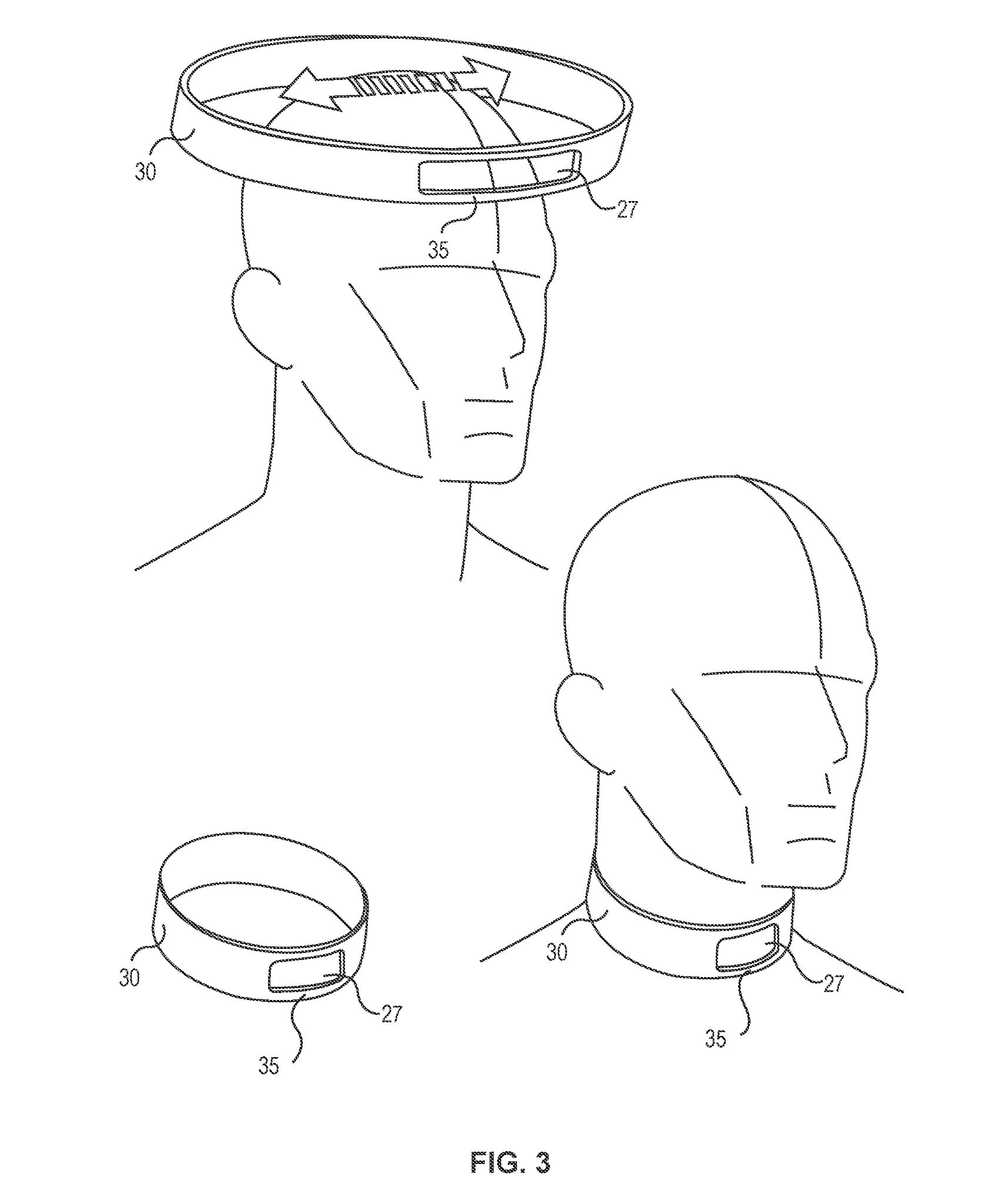
US20190297966A1 titled “Methods And Devices To Reduce Damaging Effects Of Concussive Or Blast Forces On a Subject” assigned to Q30 Sports Science describes devices for mitigating traumatic brain injury, injury to an ocular structure, or injury to the inner ear of a subject by applying pressure to one or more neck veins before and during an injurious event. The device is a partially circumferential collar device sized to be worn around a human neck and open at the laryngeal prominence, wherein the collar is adapted to apply inward pressure on the neck when worn. It includes at least one pair of pressure pad modules that are adapted to be reversibly attached to the collar device.
What can we expect in 2021?
We can expect the increase in COVID-related approvals to continue into 2021. One area that can face increased scrutiny in the future is software as medical devices (SaMDs), products that are currently classified as low-risk devices. The director of CDRH called 2021 a “reset” as it looks to manage both COVID-19 and other projects4. FDA’s near-term priorities will be addressing the next round of the medical device user fees program (MDUFA V), applying regulatory flexibilities used during the pandemic to traditional processes, and launching or piloting multiple programs such as the Safer Technologies Program (STeP).
1The Interplay between FDA and Patent Law: Infusing Organizational Knowledge for Medical Device Companies; https://open.mitchellhamline.edu/cgi/viewcontent.cgi?article=1516&context=wmlr
2https://www.fda.gov/news-events/fda-voices/year-pandemic-how-fdas-center-devices-and-radiological-health-prioritizing-its-workload-and-looking
4https://www.medtechdive.com/news/Shuren-outlines-2021-priorities-after-2020-COVID-disruption/591587/




