Die U.S. Food and Drug Administration (FDA) hat bei unserem Kampf gegen COVID-19 eine Vorreiterrolle gespielt. Unmittelbar nach dem Ausbruch der Pandemie leitete die FDA mehrere Maßnahmen zur Bekämpfung der Pandemie ein, z. B. die Gewährung eines erweiterten Zugangs und neuer Zulassungen. Es ist verständlich, dass die meisten der COVID-19-bezogenen Zulassungen im Rahmen der Notfallzulassung (Emergency Use Authorisation, EUA) und nicht im Rahmen des üblichen behördlichen Zulassungsverfahrens erteilt wurden. Solche Zulassungen haben dazu beigetragen, dass Beschäftigte im Gesundheitswesen Zugang zu wichtigen COVID-19-Medizinprodukten wie In-vitro-Diagnosekits, Point-of-Care-Geräten usw. erhalten.
Die Anmeldung von geistigem Eigentum (z. B. Patenten) und die Einreichung von Zulassungsanträgen sind eng miteinander verknüpft. Patentanmeldungen gehen in der Regel den Zulassungsanträgen voraus und zielen darauf ab, breite Aspekte der Technologie abzudecken. Zulassungsanträge, die Anmeldungen von geistigem Eigentum nicht vollständig berücksichtigen, können potenzielle Folgen hinsichtlich der Patentierbarkeit einer Anmeldung und ihrer Gültigkeit/Durchsetzbarkeit haben, die zu Verletzungsverfahren führen könnten1. Wir untersuchen die jüngsten behördlichen Zulassungen beim Center for Devices and Radiological Health (CDRH) der FDA sowie ausgewählte Patentveröffentlichungen.
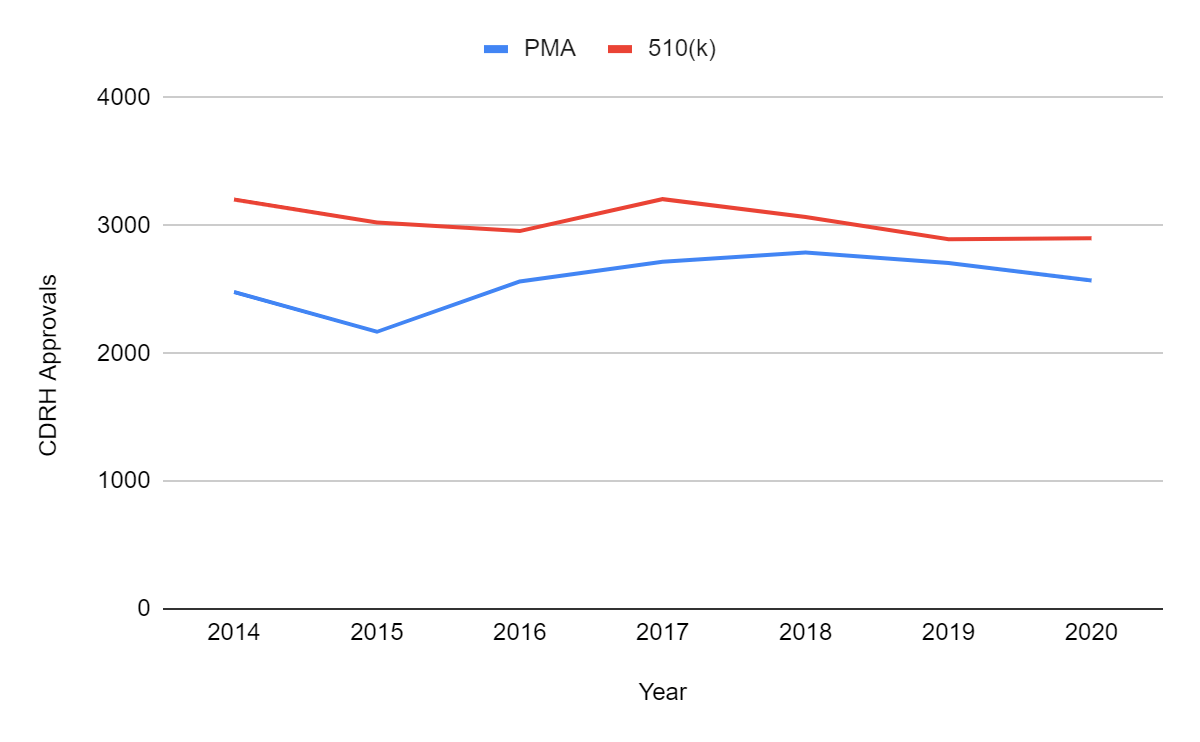
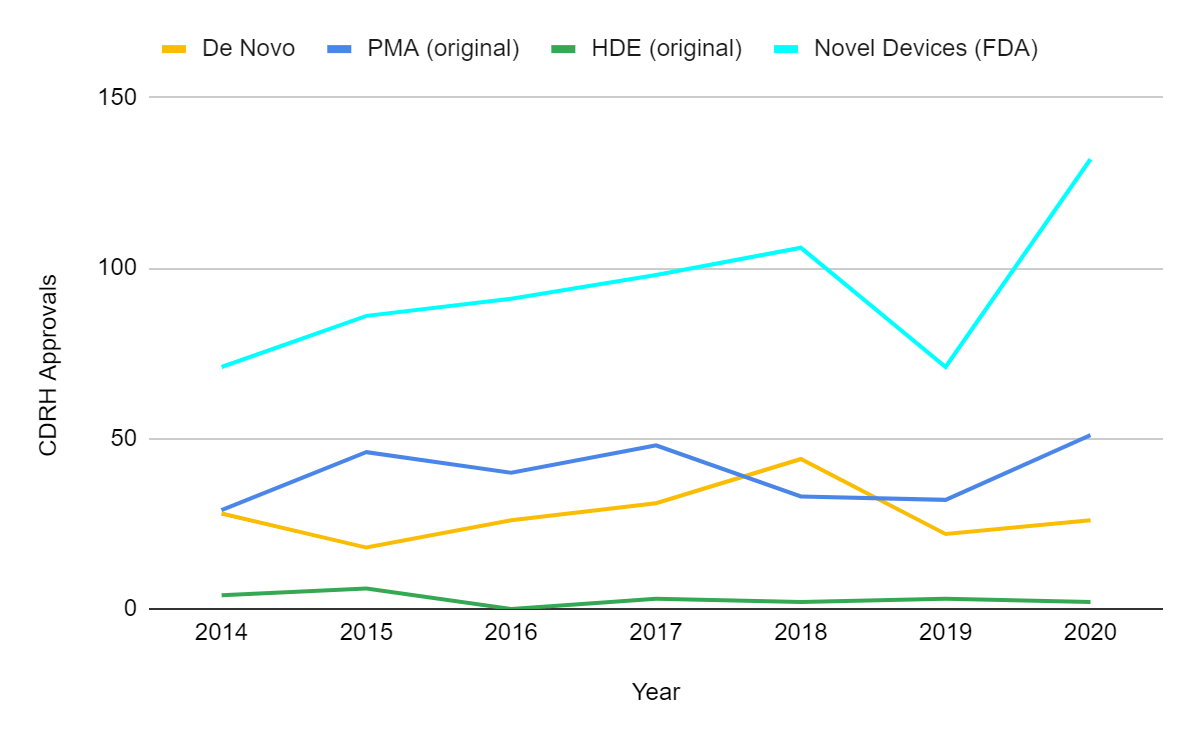
Aus den obigen Ausführungen geht hervor, dass die FDA-Zulassungen im Jahr 2020 trotz COVID-19 nicht zurückgegangen sind. Es gab keine größeren Auswirkungen auf die beiden vom Center for Devices and Radiological Health (CDRH) verwalteten Hauptpfade: Zulassungen vor dem Inverkehrbringen (PMA) und 510(k)-Zulassungen vor dem Inverkehrbringen. Im Jahr 2020 wurden 26 Geräte de novo und 2 Geräte im Rahmen der humanitären Ausnahmeregelung (HDE) zugelassen, die anderen vom CDRH überwachten Verfahren. Bis zum 1. Mai 2021 gab es 742 PMA-Zulassungen, 1039 510(k)-Freigaben, 12 De-novo-Klassifizierungen und 1 HDE-Genehmigung, was darauf hindeutet, dass sich der stabile Trend bis 2021 fortsetzen wird. Dies ist eine beeindruckende Leistung der FDA, wenn man bedenkt, dass alle jüngsten Zulassungen für COVID-19 im Rahmen von Notfallzulassungen (EUAs) erfolgten.
Bemerkenswert ist, dass die FDA im Jahr 2020 eine Rekordzahl von 132 neuartigen Medizinprodukten zugelassen hat und damit die 40-Jahres-Höchstmarke aus dem Jahr 2018 übertroffen hat. Die FDA betrachtet Anträge im Rahmen der PMA, des HDE, des De-Novo-Pfads und eine Untergruppe von 510(k)-Genehmigungen und EUA-Zulassungen als "neuartige" Produkte2. Dies liegt daran, dass die Definition der FDA für "neuartig" oder innovativ nicht einfach "neu" bedeutet. Das Produkt muss einen ungedeckten Bedarf decken oder sicherer oder wirksamer sein als derzeit verfügbare Alternativen.
Genehmigung zur Verwendung im Notfall (EUA)
Als Reaktion auf die Pandemie erteilte die FDA im Jahr 2020 625 EUAs für Medizinprodukte. Zu diesen Geräten gehörten persönliche Schutzausrüstungen, Dekontaminationsgeräte, Beatmungsgeräte, In-vitro-Diagnostika, Patientenüberwachungsgeräte, Geräte zur Unterstützung der Atmung, tragbare oder ferngesteuerte Überwachungsgeräte, Infusionspumpen, Hämodialysegeräte und andere medizinische Geräte3.

Bildquelle: © Cepheid
Der COVID-19-Diagnosetest von Cepheid für den Point-of-Care-Bereich (POC) - der Cepheid Xpert Xpress SARS-CoV-2-Test - war beispielsweise das erste POC-Diagnosegerät, das im März 2020 von der EUA zugelassen wurde. Der Test wird auf der molekularen Schnelldiagnoseplattform GeneXpert System durchgeführt.
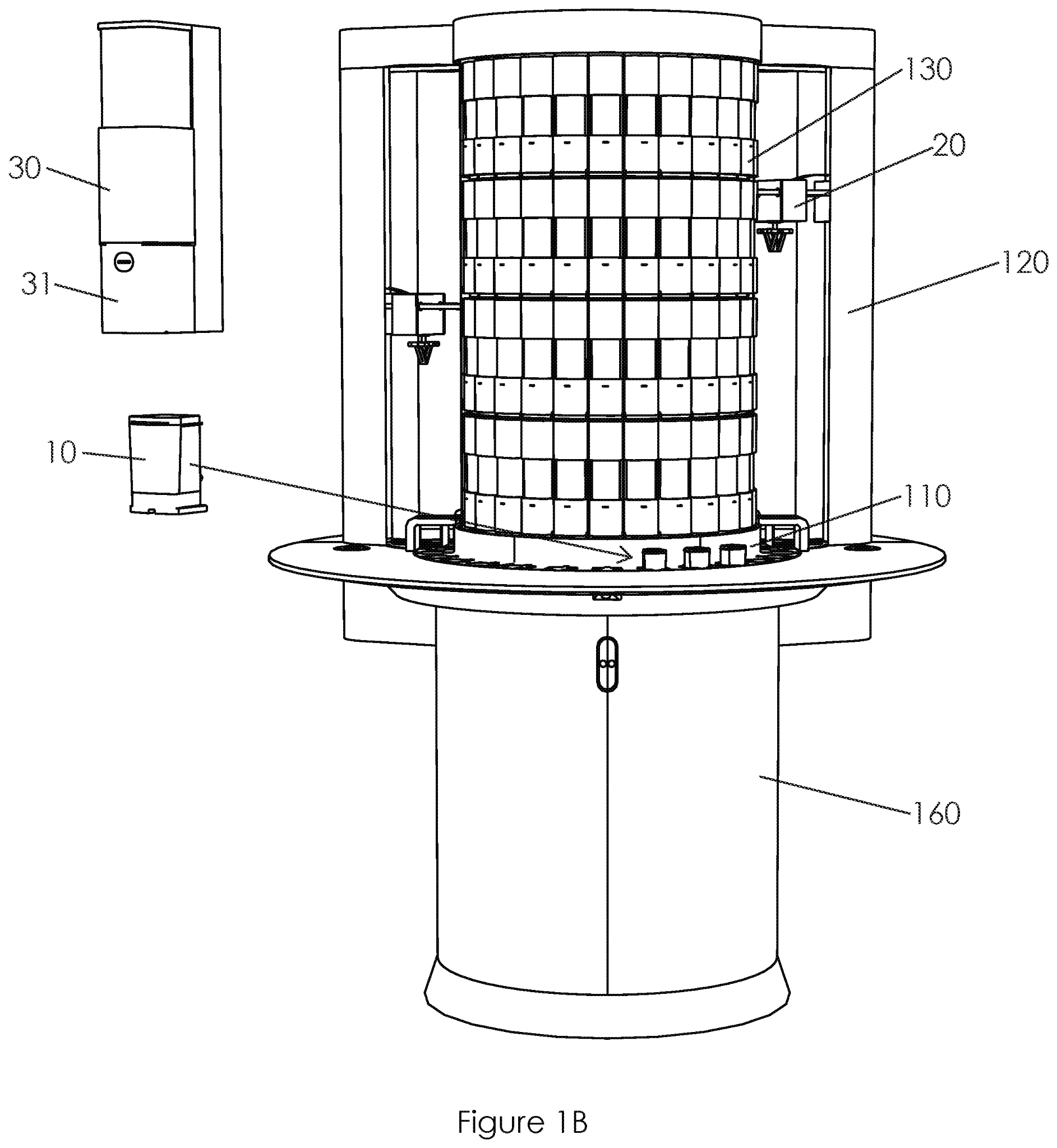
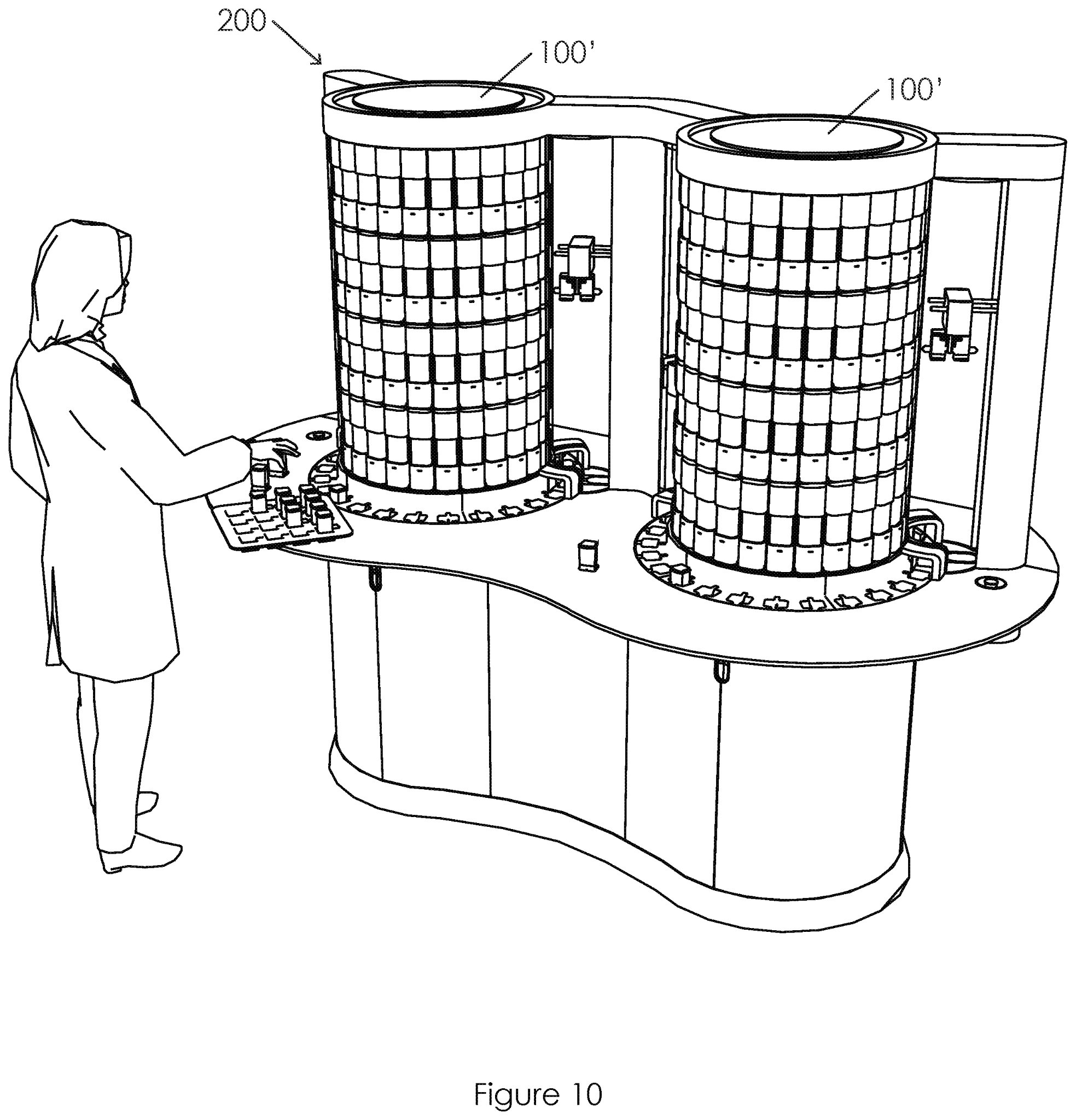
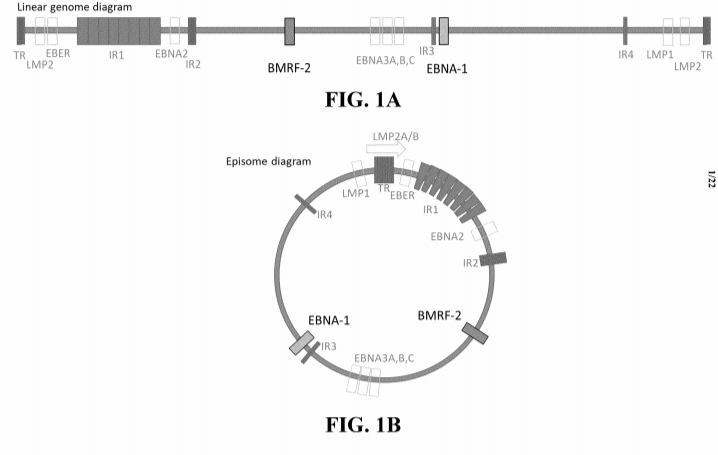
US20210072269A1 mit dem Titel "Sample Processing Module Array Handling System and Methods" (Probenverarbeitungsmodul-Array-Handhabungssystem und -Verfahren), das Cepheid mit Priorität vom 18. November 2016 zugewiesen wurde, beansprucht ein Verfahren zur Handhabung eines Hochdurchsatzsystems, das große Mengen an biologischen Proben verarbeitet. Die Proben in einer diagnostischen Assay-Kassette (10) werden durch Bewegung zwischen dem Lader (20) und der Array-Trägerbaugruppe für eine effiziente Verarbeitung platziert. Insbesondere scheint die Offenlegung das Problem des Bedarfs an einem System mit hohem Durchsatz zu lösen, das in bestehenden Einrichtungen installiert werden kann. Bei dem System handelt es sich um ein auf Kartuschen basierendes System für die Nukleinsäureamplifikation zur Durchführung diagnostischer Schnelltests. Bestehende Systeme sind groß und teuer, während kleinere Systeme keine Hochdurchsatzsysteme sind, was in einer kritischen Situation wie der Pandemie zu inakzeptablen Verzögerungen führt.
Notifizierung vor dem Inverkehrbringen - 510(k)
Die meisten Produkte der Klasse I und einige der Klasse II sind von einer 510(k)-Anmeldung befreit, wenn sie ein geringes bis mittleres Risiko für den Patienten darstellen. Die 510(k)-Notifizierung vor dem Inverkehrbringen für Produkte mit geringem bis mittlerem Risiko ist der am häufigsten genutzte Weg, der zu über 50 % aller Zulassungen beim CDRH beiträgt. Um eine 510(k)-Zulassung zu erhalten, muss der Antragsteller nachweisen, dass das neue Produkt im Wesentlichen gleichwertig mit einem Prädikatsprodukt ist. Ein Prädikatprodukt ist ein bestehendes Medizinprodukt, das in den USA legal vermarktet werden darf und als Vergleichspunkt für die Feststellung der Gleichwertigkeit dient. Das Prädikatsprodukt kann aus dem eigenen Produktportfolio oder aus dem Portfolio eines Dritten stammen.

Bildquelle: Roche
Roche Molecular Systems erhielt am 29. Januar 2021 eine 510(k)-Zulassung für einen Test zum Nachweis des BK-Virus, der auf der cobas 6800/8800 Plattform durchgeführt wird(K203220). Die Freigabe gilt für die Quantifizierung von BK-Virus-DNA aus menschlichem Plasma/Urin und ähnelt in ihren Eigenschaften dem Prädikatstest(K202215; 2. September 2020), der seinerseits seine Freigabe auf sein Prädikatstest(DEN200015; 30. Juli 2020) für die Quantifizierung von Epstein-Barr-Virus (Ebv)-DNA zurückführt. Roche erhielt außerdem weitere Zulassungen und Genehmigungen für seine Cobas-Plattform.
Interessanterweise beansprucht eine kürzlich von Roche eingereichte PCT-Anmeldung WO2021013972A1 mit dem Titel "Compositions and Methods for Detection of Epstein Barr Virus (EBV)" (Zusammensetzungen und Methoden zum Nachweis des Epstein-Barr-Virus (EBV)) mit Priorität vom 25. Juli 2019 ein Verfahren zum schnellen Nachweis des Epstein-Barr-Virus in einer Probe durch eine PCR-basierte Methode, die einen Amplifikationsschritt, einen Hybridisierungsschritt und einen Nachweisschritt umfasst. Darin werden Kits auf der Basis von Primern und Sonden für den Nachweis des Virus und Kits bereitgestellt.
Zulassungen vor der Markteinführung - PMA
Die PMA ist der strengste Weg für die Marktzulassung von Medizinprodukten. Produkte, die eine PMA erfordern, sind Produkte der Klasse III, die ein erhebliches Krankheits- oder Verletzungsrisiko darstellen, oder Produkte, die nicht für das 510(k)-Verfahren in Frage kommen. Solche Produkte der Klasse III sind oft neuartig und können auf dem 510(k)-Weg nicht als Prädikatsprodukte anerkannt werden. Die Zulassung vor dem Inverkehrbringen basiert auf der Feststellung der FDA, dass der Antrag genügend gültige wissenschaftliche Beweise enthält, wie z. B. Daten aus klinischen Studien, um zu gewährleisten, dass das Produkt sicher und wirksam ist. Aufgrund der strengen Anforderungen kann die PMA-Zulassung länger als 6 Monate dauern und ist kostspielig.
Die ersten PMA-Zulassungen im Jahr 2020 betrafen hauptsächlich den Bereich der Diagnostik - ein Trend, der sich auch 2021 fortsetzt. So erhielt der Hauptantragsteller DiaSorin PMAs für diagnostische Tests auf seiner Liaison-Plattform zum Nachweis von Hepatitis-B-, Hepatitis-C- und HIV-Antikörpern im Blut eines Patienten. Die anderen wichtigen therapeutischen Bereiche waren Kardiologie, Augenheilkunde, Orthopädie, Neurologie und Urologie. Roche, Philips, Ventana Medical Systems, Medtronic, Abbott, Boston Scientific und Foundation Medicine waren weitere namhafte Bewerber.
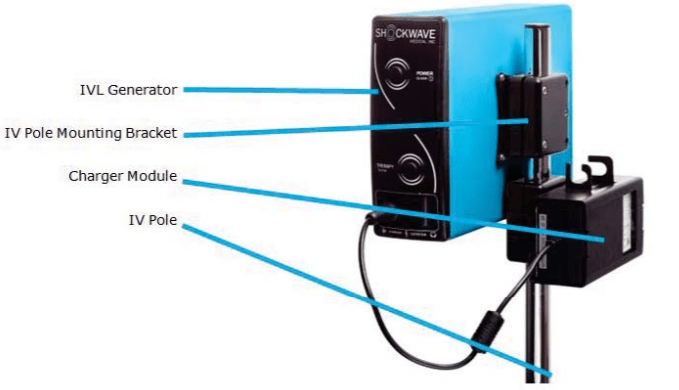


Bildquelle: FDA
Shockwave Medical erhielt am 12. Februar 2021 die PMA-Zulassung im Rahmen des Breakthrough Device Programms für sein Shockwave Intravascular Lithotripsy (IVL) System mit dem Shockwave C2 Coronary Intravascular Lithotripsy (IVL) Katheter. Das koronare IVL-System wurde entwickelt, um die Stent-Ergebnisse zu verbessern, indem es die kalziumauflösende Wirkung der Lithotripsie vor der Ballondilatation bei niedrigem Druck ermöglicht. Es besteht aus einem IVL-Ballonkatheter mit zwei integrierten Lithotripsie-Emitterpaaren, einem Lithotripsie-Generator und einem Anschlusskabel. Der NCT03595176 untersucht die Sicherheit und Wirksamkeit des Systems bei de novo, verkalkten, stenotischen Koronararterien vor dem Stenting.
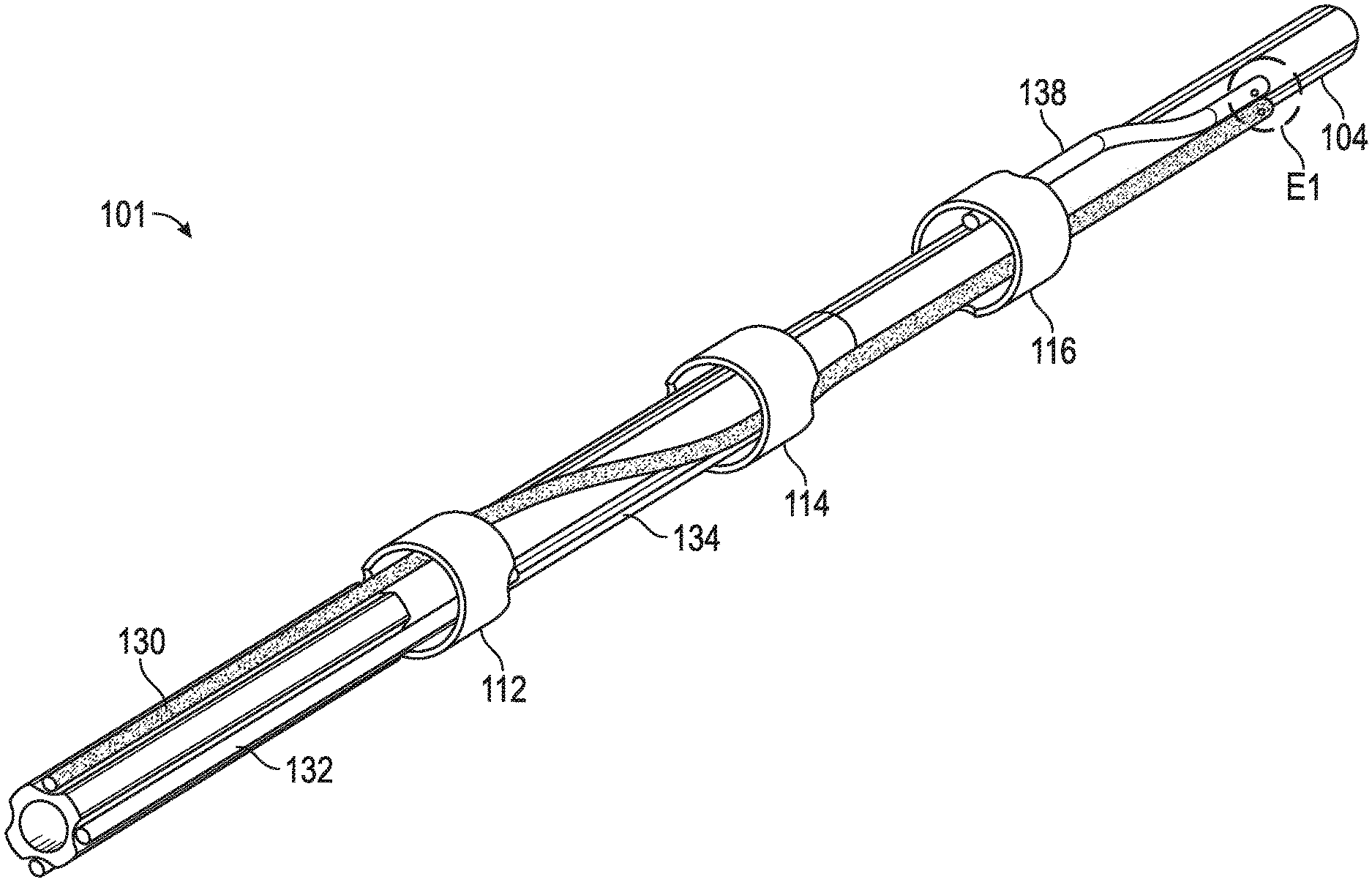
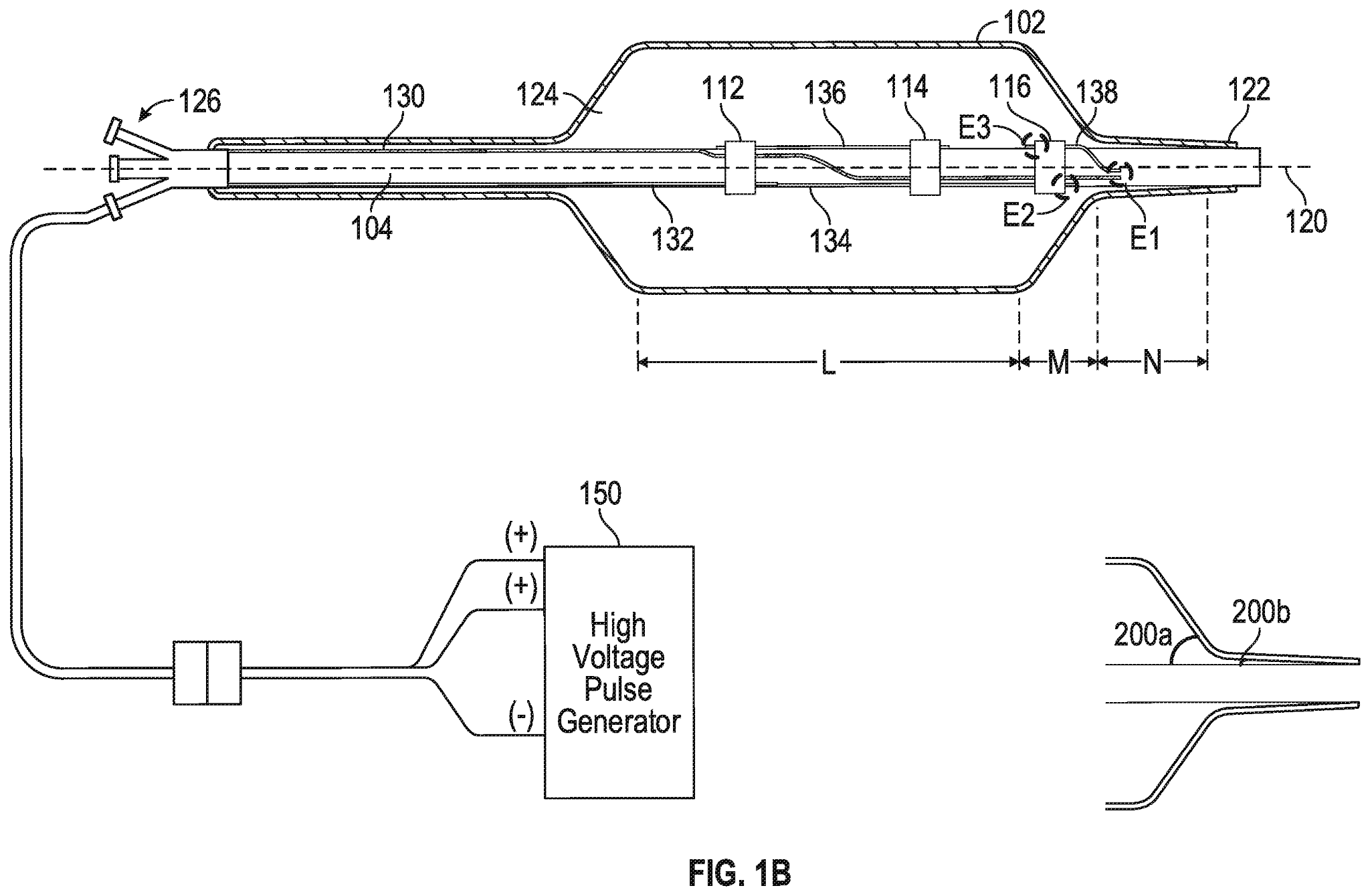
Eine kürzlich eingereichte Patentanmeldung von Shockwave Medical - US20210085383A1 - mit dem Titel "Low Profile Electrodes For a Shock Wave Catheter" (Niedrigprofil-Elektroden für einen Stoßwellenkatheter), die Priorität bis zum 24. September 2019 beansprucht, beschreibt ein System zur Behandlung enger, schwer zu durchdringender verkalkter Läsionen, bei dem ein Angioplastie-Ballon verwendet wird, um die Läsionen zu erweitern und Stoßwellen zur Wiederherstellung des normalen Blutflusses in der Arterie eines Patienten zu erzeugen. Eine solche Vorrichtung umfasst ein längliches Rohr (104) und einen Ballon (102), der in Umfangsrichtung um das Rohr gewickelt und an einem distalen Ende des Rohrs versiegelt ist. Während der Behandlung wird die Vorrichtung in das Gefäßsystem des Patienten eingeführt und der Ballon mit einer leitfähigen Flüssigkeit aufgeblasen, so dass der Ballon an den Wänden des Gefäßsystems proximal der verkalkten Läsion befestigt wird. Der Ballon enthält mindestens einen Niedrigprofil-Emitter (E1-E3), der in der Nähe des distalen Endes des Ballons positioniert ist und aktiviert werden kann, um Stoßwellen zu erzeugen, die Verkalkungen in der Läsion aufbrechen. Nachdem das Kalzium in der engen Läsion modifiziert wurde, kann der Ballon entleert und weiter in die Läsion vorgeschoben werden, um die Behandlung fortzusetzen.
De Novo-Klassifizierung
Das "de novo"-Verfahren bietet einen alternativen Weg für die Zulassung neuartiger Medizinprodukte, für die es kein rechtmäßig vermarktetes Prädikatsprodukt gibt. Ein solcher Antrag kann gestellt werden, nachdem diese Produkte automatisch in die Klasse III eingestuft wurden und im Rahmen einer 510(k)-Anmeldung die Feststellung "nicht wesentlich gleichwertig" (NSE) erhalten haben. Alternativ kann ein solcher Antrag auch gestellt werden, ohne dass zuvor eine 510(k)-Meldung eingereicht und eine NSE-Bestimmung erhalten wurde. Ein Antrag auf De-Novo-Klassifizierung kann dazu führen, dass diese Produkte zu Vermarktungszwecken in die Klasse I oder II eingestuft werden, so dass sie als Prädikate für künftige 510(k)-Anmeldungen dienen können.
Im Jahr 2020 gab es sechsundzwanzig De-Novo-Zulassungen. Die Zulassungen betrafen die Bereiche Diagnostik (8), Gastroenterologie/ Urologie (6), Kardiologie (3) und Orthopädie (3). Zum Zeitpunkt der Abfassung dieses Artikels gab es bereits 12 De-Novo-Zulassungen im Jahr 2021.

Bildquelle: FDA
Das neurologische Gerät "Q-collar" wurde am 26. Februar 2021 im Rahmen der De-Novo-Klassifizierung zugelassen. Das Gerät ist ein externes Kompressionsgerät für die Kompression der inneren Jugularvene. Es hat die Form eines C-förmigen Kragens, der eine Kompressionskraft auf den Hals ausübt und das Blutvolumen erhöht, um die Bewegung des Gehirns im Schädelraum zu verringern, die bei Kopfstößen auftreten kann. Wenn der Q-Collar bei sportlichen Aktivitäten um den Hals getragen wird, übt er eine Druckkraft auf die inneren Jugularvenen aus, was wiederum das Blutvolumen in den Blutgefäßen des Schädels erhöht. Bei Unfällen mit stumpfem Trauma bewegt sich das Gehirn in der Regel unkontrolliert im Schädel, was als "Schwappen" bezeichnet wird. Die Vergrößerung des Blutvolumens in den Blutgefäßen durch den Q-Collar sorgt für einen festeren Sitz des Gehirns im Schädel und reduziert die Schwappbewegungen. Durch die Verringerung der Bewegung des Gehirns innerhalb des Schädelraums kann der Q-Collar zum Schutz des Gehirns vor den Auswirkungen von Kopfstößen beitragen.
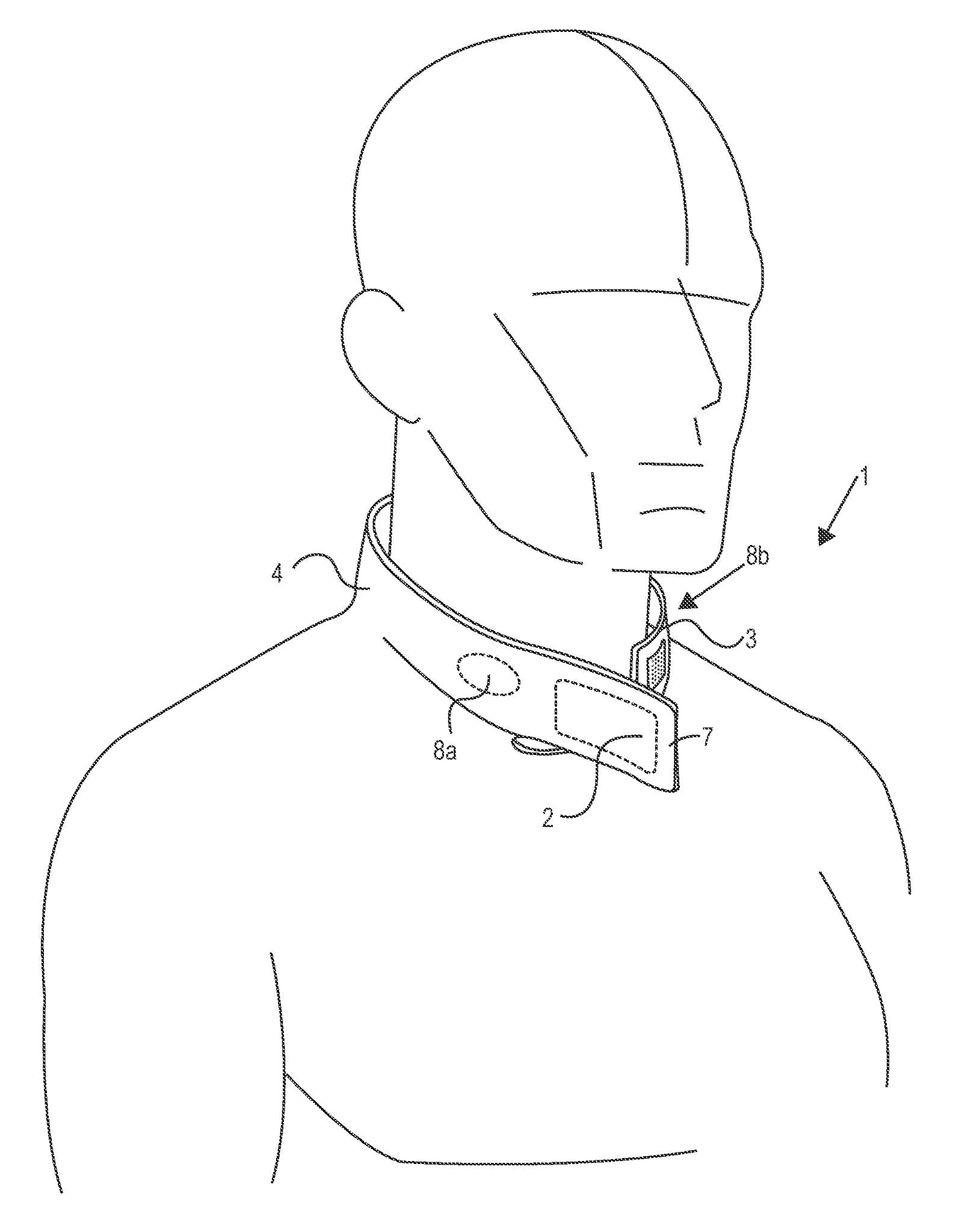
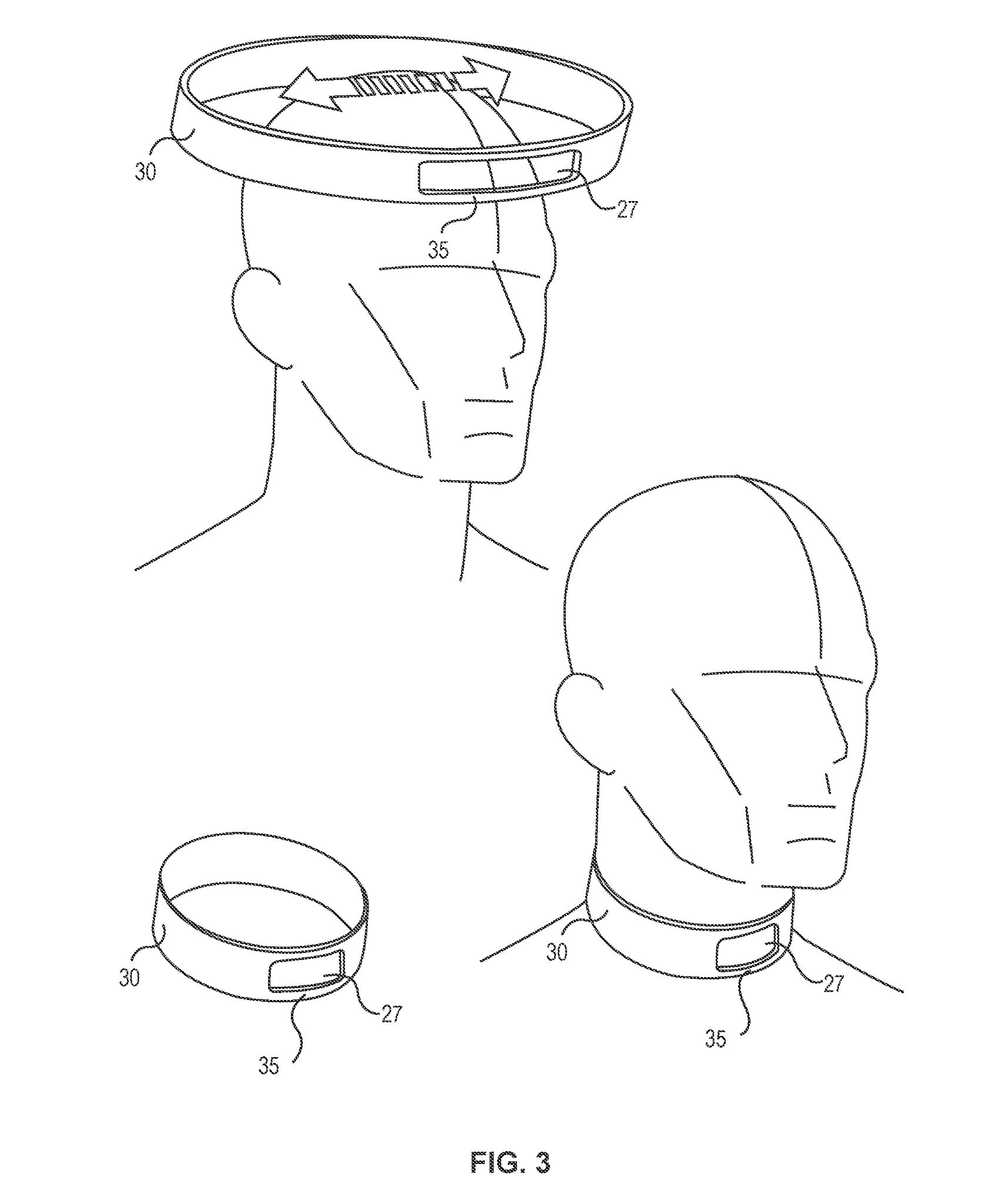
US20190297966A1 mit dem Titel "Methods And Devices To Reduce Damaging Effects Of Concussive Or Blast Forces On a Subject" (Methoden und Vorrichtungen zur Verringerung der schädigenden Auswirkungen von Erschütterungs- oder Explosionskräften auf eine Person), die Q30 Sports Science zugewiesen wurde, beschreibt Vorrichtungen zur Milderung von traumatischen Hirnverletzungen, Verletzungen einer Augenstruktur oder Verletzungen des Innenohrs einer Person, indem vor und während eines Verletzungsereignisses Druck auf eine oder mehrere Halsadern ausgeübt wird. Bei der Vorrichtung handelt es sich um eine teilweise umlaufende Kragenvorrichtung, die so bemessen ist, dass sie um einen menschlichen Hals getragen werden kann und am Kehlkopfvorsprung offen ist, wobei der Kragen so angepasst ist, dass er beim Tragen Druck nach innen auf den Hals ausübt. Sie umfasst mindestens ein Paar Druckkissenmodule, die reversibel an der Kragenvorrichtung angebracht werden können.
Was können wir im Jahr 2021 erwarten?
Es ist zu erwarten, dass sich der Anstieg der COVID-bezogenen Zulassungen bis 2021 fortsetzen wird. Ein Bereich, der in Zukunft stärker unter die Lupe genommen werden könnte, ist Software als Medizinprodukte (SaMD), Produkte, die derzeit als Produkte mit geringem Risiko eingestuft werden. Der Direktor des CDRH bezeichnete das Jahr 2021 als einen "Reset", um sowohl COVID-19 als auch andere Projekte zu verwalten4. Zu den kurzfristigen Prioritäten der FDA gehören die nächste Runde des Gebührenprogramms für Medizinprodukte (MDUFA V), die Anwendung der während der Pandemie genutzten regulatorischen Flexibilität auf herkömmliche Verfahren und die Einführung oder Erprobung mehrerer Programme wie das Safer Technologies Program (STeP).
1DasZusammenspiel zwischen FDA und Patentrecht: Vermittlung von Organisationswissen für Unternehmen der Medizintechnik; https://open.mitchellhamline.edu/cgi/viewcontent.cgi?article=1516& context=wmlr
2https://www.fda.gov/news-events/fda-voices/year-pandemic-how-fdas-center-devices-and-radiological-health-prioritizing-its-workload-and-looking
4https://www.medtechdive.com/news/Shuren-outlines-2021-priorities-after-2020-COVID-disruption/591587/




